Modeling Biological Networks
IV.1 Coordinators
IV.2 Participants
IV.3 Introduction
IV.4 Background and Significance
IV.5 Research Plan
IV.6 Specific Subprojects
- IV.6.iv Subproject 4 Modeling the Regulation and Function of Metabolism
- [ Other Subprojects ]
IV.8 Timeline
< Previous | Page 25 of 35 | Next >
IV.6.iv.d.2.ii Molecular validation:
To determine the vulnerability of the E. coli metabolic network, we plan to identify and characterize changes in the network structure upon the removal of a single enzyme, E1. Depending on the nature of E1 and the properties of the components (nodes) participating in the reaction it catalyzes, deletion can have different effects. For example, in Figure IV.9 a, if the main product X of the reaction is an authority, i.e., a number of other reactions produce it simultaneously, removing E1 will have practically no effect, since other reactions take up its role. However, if product X is a hub (Figure IV.9 b) and participates in numerous reactions as an educt, the removal of E1 can disrupt a large portion of the metabolic network. Using the available metabolic maps, we plan to characterize the effect of the removal of all individual metabolic enzymes on the substrate connectivity of the metabolic network.
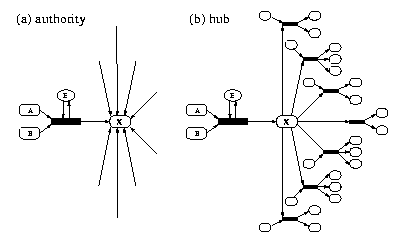
| Fig. IV.9. Authorities and hubs in a metabolic network. (a) An authority is a node with many incoming links, i.e., many independent reactions can produce it. (b) A hub is a node with many outgoing links, i.e., it is an educt for many reactions. Nodes (e.g. H2O) can simultaneously be authorities and hubs. |
IV.6.iv.d.2.iii Experimental validation:
To further investigate the effect of eliminating a specific enzyme in different cells, we will correlate our theoretical predictions on the qualitative importance (i.e., essential vs. non-essential) of enzymes with systematic gene disruption data. We will thus be able to distill the topologic characteristics of fatal enzyme removal, providing useful information about the importance or irrelevance of certain disruptions. For example, we might find that an enzyme, though it affects only a few reactions, might be essential because it affects an important set of reactions generating key metabolic intermediates (i.e., hubs). Another enzyme, whose elimination potentially could disrupt a larger fraction of the network, might prove non-essential because it catalyzes the synthesis of components that are less critical for the cell's immediate survival (i.e., authorities).
Initially we will consider available data on yeast, S. cerevisiae. Two recent studies achieved precise deletion of one of ~2000 ORFs of the S. cerevisiae genome (~ one-third of the ORFs in the genome) (Winzeler et al., 1999; Ross-Macdonald et al., 1999), identifying ~17% of these ORFs as essential for viability, with an additional ~40% showing quantitative growth defects (Winzeler et al., 1999). Since mutated yeast strains can propagate either as diploids or haploids, switching from diploid to haploid accurately distinguishes essential from non-essential genes. We can compare the list of essential and non-essential enzymes in these databases with our theoretical predictions of the importance of an enzyme to the integrity of the metabolic network.
Once we firmly establish our mathematical approaches, we will pursue a similar analysis in E. coli. F. Blattner and G. Church, a consortium led by T. Mizuno and K. Isono in Japan, and a consortium in Europe led by M. Masters are independently conducting systematic mutagenesis studies in E. coli. The literature and on-line databases contain substantial data on individual mutants (e.g., the PEC database: http://www.shigen.nig.ac.jp/ecoli/pec/index.html). The recent complete sequencing of the 0157:H7 enterohemorrhagic E. coli strain (Perna et al., 2001) identified distinct differences with the genome of E. coli K12 (our model), suggesting possible differences in the organization of genetic regulatory networks between the two strains. Thus, our manually curated dataset will initially be restricted to individual mutants in E. coli K12, but our analytical approach will incorporate other data as they become available as well.
Molecular validation in E coli: The predictions generated by our computer simulations above will need to be experimentally verified (proof-of-principle). Based on theoretical predictions, we will generate several lists of enzymes whose individual or combined removal, -we predict-, will lead to the collapse of the metabolic network when E. coli is grown on defined media, leading to death or very severe growth defects. We will test the viability, growth rate, and metabolic profile of selected single mutants to confirm that in themselves these mutations have no dramatic effect on growth, and test double mutants as well, to compare with the theoretical predictions. The gene manipulation experiments will be the primary responsibility of members of the B. L. Wanner research group (Purdue). Members of the Z. N. Oltvai and B. L. Wanner research groups will carry out the gene expression profiling experiments jointly.
Dynamical validation: The predictions of our models will be directly tested by microarray analysis of the Escherichia coli transcriptome. In collaboration with Integrated Genomics we plan to create a cDNA chip in which all proven and many predicted open reading frame of the E. coli genome are deposited in triplicate. The temporal change in the global transcriptional activity in E. coli will be monitored by collecting a serial of time points during metabolic shift experiments e.g., glucose to galactose switch. The results will be combined with the genome-wide, comprehensive experimental assessment of the genes necessary for robust growth of E. coli in a rich, tryptone-based media. Gene essentiality will be determined by genetic foot-printing of E. coli MG1655 based on Tn5-transposome mutagenesis (Gerdes, 2002), extending this to the whole-genome using the standardized growth conditions. Subsequently, we then plan to project individual gene assessments onto a whole-cell functional reconstruction including metabolic and non-metabolic subsystems and analyze the distribution of conditionally essential and dispensable E. coli genes within functional groups with respect to the occurrence of putative orthologs across a broad range of diverse bacterial genomes.
Protein Interactions: While functional experiments examining the effects of multiple deletions will be an important validation of the predictions, it may be informative to directly identify interactions between proteins. For example, metabolic collapse from the deletion of two enzymes could result from deletion of proteins which share a common substrate yet do not physically interact. An alternative scenario, however, is that the two proteins directly interact in a regulatory fashion. Often such interactions may be identified in the literature. In cases where an interaction is unknown, however, direct experimental examination using purified proteins will be possible using Biacore or surface plasmon resonance (SPR) technology. In this technique one protein is immobilized on a hydrophilic sensor surface in the SPR spectrometer and the second protein (the putative binding partner) is flowed over the surface. An interaction between the proteins is detected as a signal that is proportional to the increase in mass on the surface. Although quantification in terms of affinity and kinetics are possible, at this level we will primarily be interested in whether an interaction is detected. However, when testing more complex predictions, we may be interested in relative affinities between multiple proteins. Assaying relative affinities between multiple partners is one of the primary advantages of Biacore technology. Other advantages are that the technique is relatively high-throughput and multiple interactions can be tested simultaneously. These experiments will primarily be the responsibility of the laboratory of B. Baker (University of Notre Dame). This lab is equipped with a Biacore 3000 SPR instrument. The Biacore 3000 is optimized for high sensitivity and maximum throughput.